
Research Themes

Photophysics of organic materials mediated by intermolecular interactions
Porous materials offer a combination of high surface area and thermal stability that has driven their adoption for applications ranging from gas storage and separation to heterogeneous catalysis. These materials also offer advantages as a platform for photophysical and photochemical applications. We are using molecular simulations to develop a mechanistic understanding of how the complementary strategies of chromophore embedding and encapsulation can favorably modify the electronic properties of photoactive porous materials.
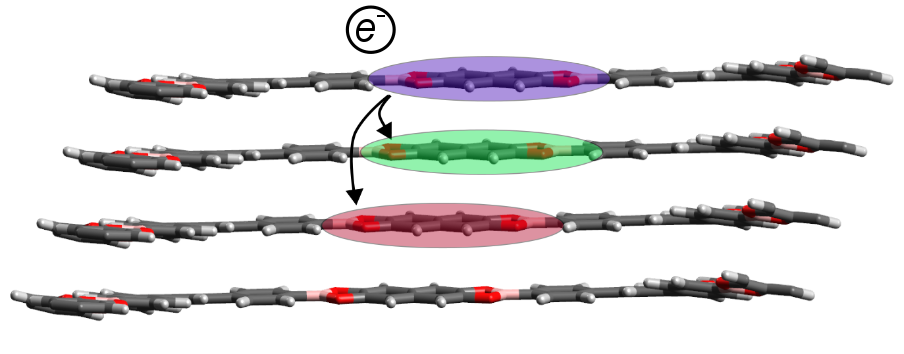
Structural and electronic properties of covalent organic frameworks
Covalent organic frameworks (COFs) are a class of ordered, porous materials that can assemble into 2D or 3D networks depending on their chemical building blocks. We are studying how the light-absorbing and electronic properties of organic chromophores can be modulated by embedding of the chromophore within a COF. Through electronic structure simulations based on constrained density functional theory, we seek to determine whether COFs can be engineered to support singlet fission (SF), the creation of two triplet excitons from one singlet exciton. In the active layer of an organic photovoltaic cell, materials with this property could dramatically enhance the efficiency of converting light to electricity.
Modulation of charge transfer state lifetimes by encapsulation
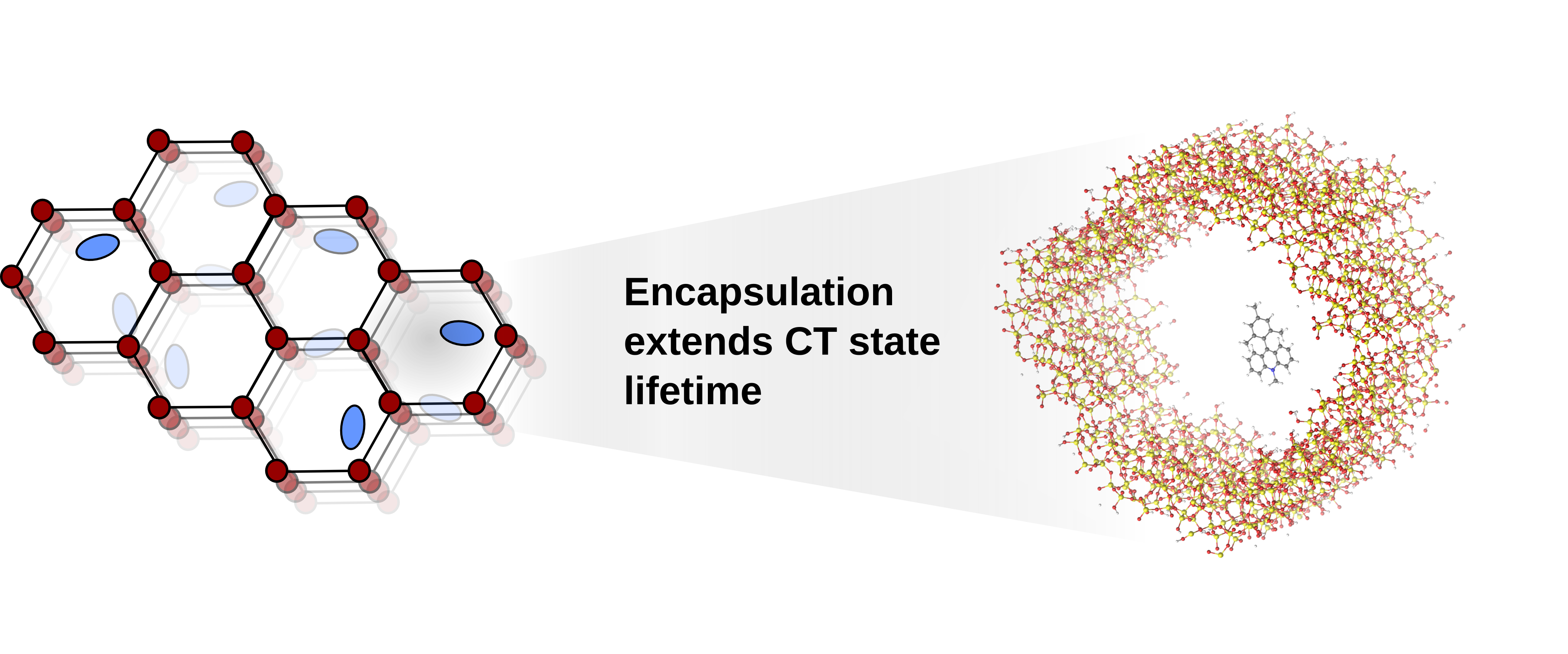
The efficiency of chemical processes mediated by electronically excited states is limited by the typically very short lifetime (picoseconds or even femtoseconds) of the excited state. Recent experiments have suggested that encapsulation of donor-acceptor chromophores in mesoporous materials can dramatically extend the lifetime of charge-transfer excited states. We have developed a multiscale simulation approach that combines Monte Carlo and molecular dynamics sampling to simultaneously explore chromophore-pore interactions alongside internal degrees of freedom on the chromophore. These simulations are helping us to understand how changes to a chromophore's environment may alter its photophysical properties.
Efficient computational tools for simulating electronic excitations and photostability
To enable high-throughput computational screening of photoactive materials, our method development efforts focus primarily on tools for rapid characterization of low-lying electronically excited states. We have introduced a simple time-independent approach to excited states, based on the Δ-self-consistent-field approach, within the semi-empirical density functional tight binding (DFTB) formalism. Benchmarks of excited state geometries and properties, including vibrational frequencies and Stokes shifts, have demonstrated the ability of ∆DFTB to efficiently screen materials for these properties.
Photochemistry and singlet oxygen sensitization of photodynamic therapy agents
For the selective destruction of biological tissue, photodynamic therapy employs chromophores that are excited by light to generate singlet oxygen or reactive oxygen species. The electronic properties and relaxation pathways of such chromophores are essential to their further optimization, but fast methods for molecular dynamics sampling of such dyes in their electronically excited states are limited. We have undertaken a comparative analysis of the lowest singlet excited state potential energy surface (PES) of the BODIPY chromophore obtained from time-dependent density functional theory (TDDFT) and from the restricted open-shell Kohn-Sham (ROKS) approach. The findings from this investigation are guiding our studies of the interaction of BODIPY dyes with molecular oxygen through DFT and DFTB simulations.
Balanced ground and excited state characterization of solar thermal fuels
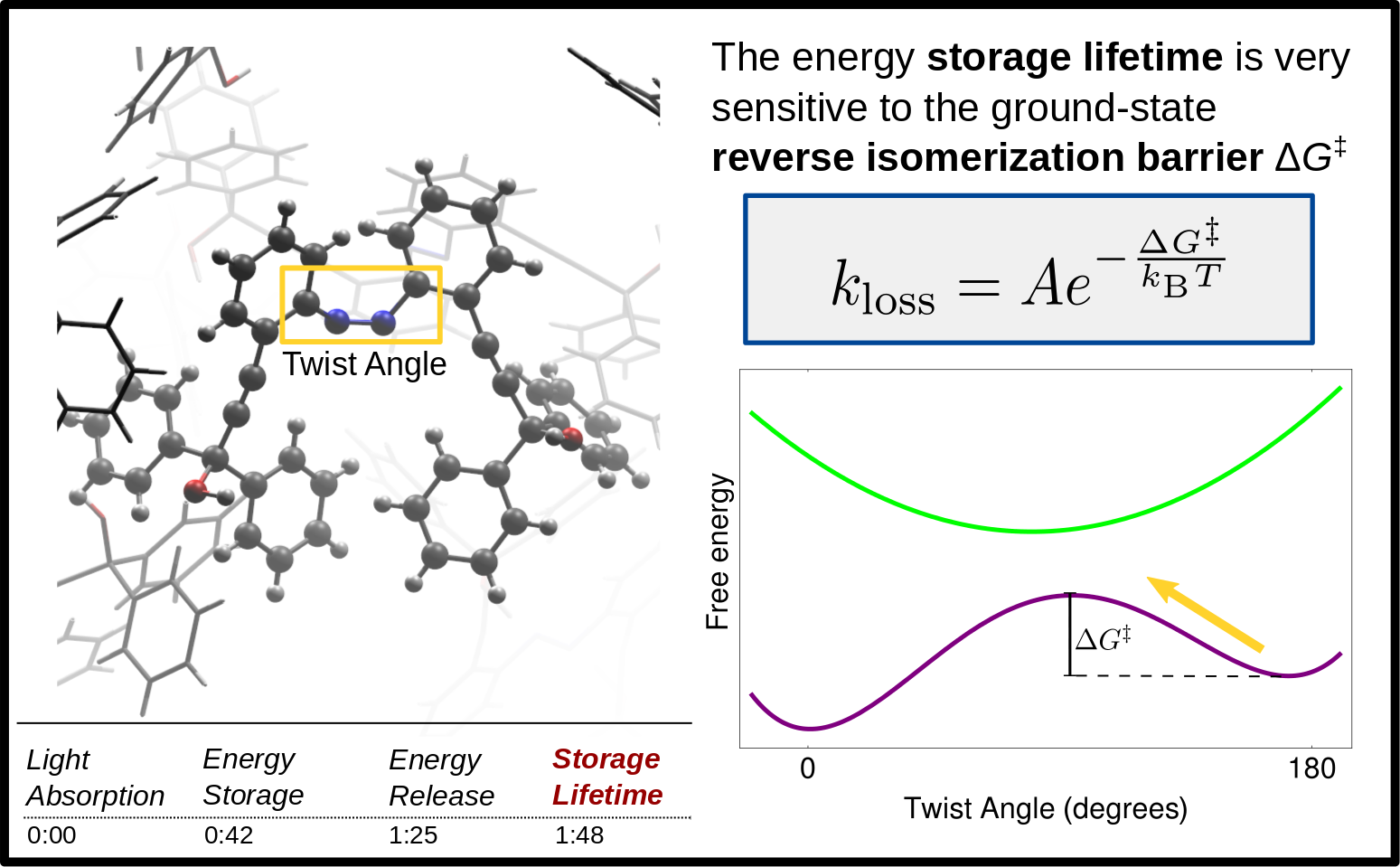
Liquid solar thermal fuels (STFs) store chemical potential energy in molecular strain induced by excited-state photoisomerization in sunlight. A number of photoswitching scaffolds have been identified for the development of STFs, but solar spectrum matching and energy density remain challenging properties to optimize. We are developing a computational screening protocol that employs both ground and excited state DFTB to search for candidate STFs that simultaneously meet several key structural and energetic criteria.
Research Sponsors
We gratefully acknowledge financial support of this research program from the following organizations.
- American Chemical Society Petroleum Research Fund
- National Science Foundation (CHE-1664674, DMR-1848067)
- Research Corporation for Science Advancement
- Western Washington University Research and Sponsored Programs
- WWU Advanced Materials Science and Engineering Center
We also thank the following organizations for supporting initiatives that have enabled student and visitor participation in our research.
- Snohomish Public Utility District
- Royal Golden Jubilee (RGJ) Fellowship Program